In this module we obtain accurate structures for the compound of interest in three different media: gas-phase, water and octanol.
For each compound we consider:
- The existence of E/Z isomers and possible stereoisomers
- Presence of different tautomers
- A systematic search of the conformational space.
Structures of the most stable tautomers and conformers are obtained individually to reach 99% of the Boltzmann distribution.
For some molecules with great conformational freedom, the number of studied conformations can be very high (hundreds). Note that the ensemble will also depend on the environment, therefore we have separate results for gas phase, water and octanol solvents.
- In a first step the conformational is done using the CREST software (Pracht, P.; Bohle, F.; Grimme, S. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192) which includes a metadynamics (MTD) and Genetic z-matrix Crossing (GC) steps.
- In a second step we use the the CENSO (Commandline ENergetic SOrting) software (Grimme, S.; Bohle, F.; Hansen, A.; Pracht, P.; Spicher, S.; Stahn, M. J. Phys. Chem. A 2021, 125 (19), 4039–4054) under the following scheme.
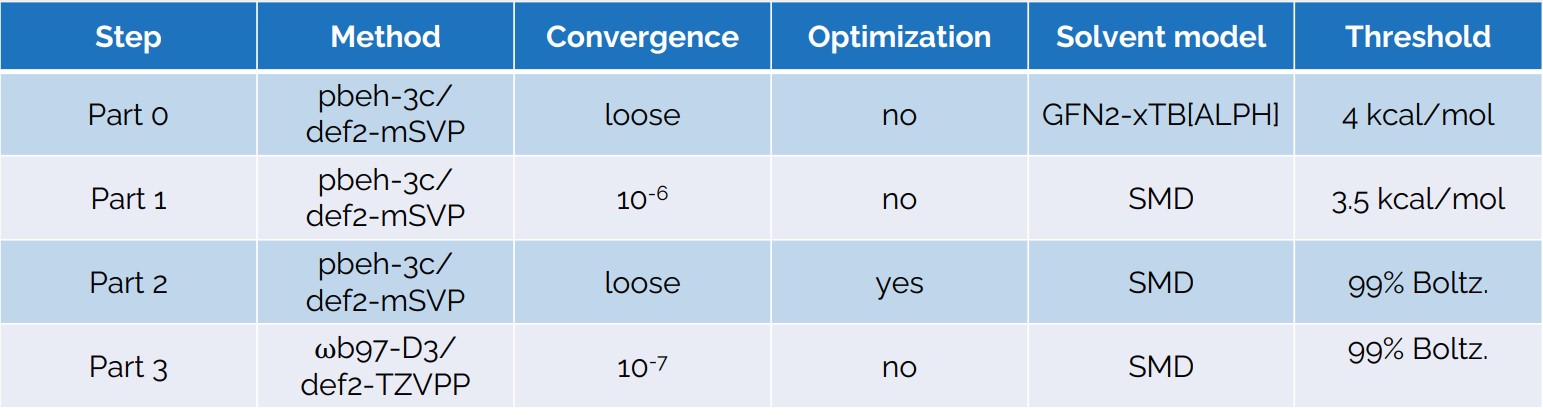
- Results from the final step using ORCA software are given in the SEPIA app.
Once conformers are generated, we perform high-level QM density functional theory calculations using Gaussian 16 software suite to calculate accurate molecular properties.
These calculations are crucial since they are used to feed the system for obtaining predictive models.
To obtain the final properties of each conformer we have used the Gaussian software package (Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian~16 Revision C.01, 2016.)
The following calculations are performed:
- Geometry optimization and frequency calculation (IR spectra) at B3LYP-D3/6-311+G(d,p) level of theory.
- TD-DFT calculation at B3LYP-D3/6-311+G(d,p) to obtain the first 10 excitations and the UV-Vis spectra.
- Single point calculation at B3LYP-D3/6-311+G(3df,2p) level of theory to obtain accurate energies.
- Topological analysis of the electron density.
In this module we perform molecular dynamics (MD) simulations to identify the most labile bonds and predict which are the most probable degradation products.
Our aim is to predict the fragmentation routes seen in mass spectra as well as the subproducts that depend on the environmental media.
All predicted degradation products are calculated using Modules A and B and incorporated in the database (Module D).
Work in progress
Coming soon
This module collects all the information obtained from calculations performed at Mod. B and arranges it in a structured database.
All results obtained are included in a database that is accessible through the app SEPIA and includes:
- Information of most stable conformer obtained in the final step of CENSO using ORCA and results from high level calculations using Gaussian.
- Links to full results using the IochemBD platform
The last module is responsible for the prediction of physico-chemical, environmental and ecotoxicological properties using QSAR/QSPR models.
Up to now we offer predictions for solubility in octanol and water and for octanol/water partition coefficient.
Our objective is to improve our predictions by using machine learning tools and to extend them to other physico-chemical properties and environmental and toxicological properties.
Work in progress
Coming soon